Lists and other Collections
The first part of chapter 12 of Hello World introduces lists and tuples.
There are also other types that use the square-bracket notation in
some way.
Two miscellaneous things about indices. First, you can
use -1 as the index of the last element of a
list, -2 as the second last, and so on.
Second, len() gives the size of a list or other
sequential type.
Tuples and Generators vs Lists
Tuples is a more specialized relative of lists. They look the
same, except for round brackets instead of square. The important
difference is that a tuple is immutable.
Any list can be converted to a tuple, and any tuple into a list.
Why use tuples at all? The reason lies in implementation. Tuple
operations are faster, because they don't have to allow for
mutability. Even though it doesn't matter for simple programs, Python
syntax is designed to take advantage of the immutability where
possible. Hence the distinction between lists and tuples.
One other difference is that tuples can appear implicitly, even
without the round-bracket notation. For example
x,y = y,x
exchanges two variables.
Still more specialized are generators. A generator is like a
drip-feed that gives you elements one at a time rather than a whole
list. A useful example is enumerate() which makes tuples
of list elements with their indices. The said tuples are available
one at a time, in a for loop. Try these examples:
bases = ['A','T','C','G']
for x in enumerate(bases):
print x
for i,b in enumerate(bases):
print i,b
for i in range(len(bases)):
print i,bases[i]
The three for loops are equivalent, but the ones
using enumerate are a little more readable.
Strings
Chapter 21 of Hello World is devoted to strings. But
following are some particularly useful features.
Just as with lists and tuples, you can take a single character or
a slice out of a string, using the square-bracket notation. Here's an
example of slicing.
"It's pining for the fjords."[5:-1]
A string can be split up into a list. To see how this this works,
try the following.
s = "It's pining for the fjords."
s.split()
s.split('f')
s = "It's pining for the fjords."
s.split()
Incidentally, the fragment also illustrates two syntactic features
of Python (also of some other languages, including C++ and Java).
First, the dot syntax: this is object oriented notation, meaning that
something is being done to s. Second, the empty
parentheses: if we want to split about blank, we can simply leave the
argument out.
A list of strings can be joined into a single string, as
follows.
l = s.split()
' '.join(l)
The notation here is rather odd, but it emphasizes
that join is the inverse of split.
A less drastic string operation than split is partition. In contrast
to split, it generates a tuple. The
following example will show you what it does.
s = 'I am shocked -- shocked to find \
that gambling is going on in here.'
print s.partition('shocked')
By the way, the backslash \ is a way of extending an
input line.
Another useful string operation is translate, which
gives a new string with some characters replaced. The simplest kind
of replacement is replacement with nothing, or removal, as here.
s = 'A quick brown fox jumped over the lazy dog'
print s.translate(None,'AEIOUaeiou')
With the string operation replace only one replacement can be made.
However, in contrast to translate, a substring of more than one character can be replaced.
s = 'A quick brown fox jumped over the lazy dog'
print s.replace('brown','red')
Solve this
Python
challenge. The challenge is stated as a riddle, and you will need some
lateral thinking to see what it is that you are being asked to do.
Once that is clear, you can write a program to do it. The
partition and translate operations, as well as the source code
(extension has been changed from .html to .txt) may be
useful.
hint
Proteomics
The peptide atlas
contains proteomics data on several organisms and
tissues. Comma-separated text files (csv format) with
human brain proteins
and
human plasma proteins
were obtained from the peptide atlas. The first lines of the files
contain headers. Each consecutive line contains different data for
each protein identified, starting with the biosequence name. This is a
protein identifier that protein atlas acquired from other
databases.
For this question there are some warmup
problems.
Generate lists of biosequence names for proteins
that have been found in the brain and in the plasma,
respectively. Compare these lists and generate the following console output:
Common biosequences:
<print list here>
Brain specific biosequences:
<print list here>
Plasma specific biosequences:
<print list here>
Number of common biosequences: ...
Number of brain specific biosequences: ...
Number of plasma specific biosequences: ...
Submit the program
Arrays
An array (full name numpy.ndarray) is a data type
defined by a library rather than in core Python. Arrays do not appear
in Hello World or in most other books on Python, because
their use is rather specific to scientific computing. In numerical
work, one often wants to do the same operation on many different
operations at once, and that is what arrays facilitate. When printed,
arrays look like lists without commas, but they work rather
differently.
One way to create an array is the following:
from numpy import linspace
x = linspace(0,3,7)
Try a few examples to see exactly what the arguments
to linspace() do.
Now try these.
a = linspace(-1,1,5)
a[0] = 1
b = a + a
a += 2
a *= 2
and print at each stage to see what happens. As you can see,
arrays implement vector arithmetic.
One subtlety to be aware of is illustrated by the following.
a = linspace(-1,1,5)
b = a
c = 1*a
b[0] = 26
As you can see by printing, b is just a synonym
for a whereas c is an independent copy.
An array can also be created using zeros or using array. Try these.
from numpy import zeros, array
a = zeros(6)
b = 6*[2] # b is list
b = array(b) # now b is an array
It is possible to change values within an array, but, in contrast to lists,
it is not possible to add new elements, nor to remove elements. Indexing and
slicing works the same for arrays and lists. Compared to lists,
arrays are often more suitable for carrying out calculations.
a=[5,3,7,1]
b=[9,5,5,6]
Add up the numbers in these lists (result: [14,8,12,7]). Do this without and with
using arrays. Try to make the code as short as possible.
If you want to know more about arrays, this tutorial
gives information about creating, reshaping, indexing, etc.
Dictionaries
The second part of chapter 12 introduces another Python data type: dictionaries.
The most important features are summarized here. The name 'dictionary' is slightly
misleading, because a Python dictionary has no ordering. It is like a heap of
cards, each with a word and its meaning. Thus, unlike sequences, dictionaries
are not indexed by a range of numbers. Instead, they are indexed by keys, which
are usually strings or numbers. It is best to think of a dictionary as an unordered
set of key: value pairs, with the requirement that the keys within a library are
unique.
An empty dictionary can be created by a pair of braces. A new key: value
pair can directly be added like in this example:
cdn = {}
cdn['ttt'] = 'F phenylalanine'
cdn['ttc'] = 'F phenylalanine'
cdn['tta'] = 'L leucine'
This is equivalent to:
cdn = {}
cdn['ttt'] = cdn['ttc'] = 'F phenylalanine'
cdn['tta'] = 'L leucine'
Individual elements can be accessed as cdn['ttc'] and
so on. As for lists, the ‘in’ keyword can be used for dictionaries. A key:value
pair can be removed using del:
del cdn['ttt']
The values can be of different types, such as strings or lists. If the value
is a list, functions to manipulate lists can be applied to it, like in this example:
DNA_fragments={}
DNA_fragments['mouse']=[]
DNA_fragments['mouse'].append('ACTTAAT')
DNA_fragments['mouse'].append('GCATGGC')
The genetic code
The standard genetic code is conveniently expressed in a Python
dictionary.
cdn = {}
cdn['ttt'] = cdn['ttc'] = 'F phenylalanine'
cdn['tta'] = cdn['ttg'] = 'L leucine'
cdn['tct'] = cdn['tcc'] = cdn['tca'] = cdn['tcg'] = 'S serine'
cdn['tat'] = cdn['tac'] = 'Y tyrosine'
cdn['taa'] = cdn['tag'] = ' '
cdn['tgt'] = cdn['tgc'] = 'C cysteine'
cdn['tga'] = ' '
cdn['tgg'] = 'W tryptophan'
cdn['ctt'] = cdn['ctc'] = cdn['cta'] = cdn['ctg'] = 'L leucine'
cdn['cct'] = cdn['ccc'] = cdn['cca'] = cdn['ccg'] = 'P proline'
cdn['cat'] = cdn['cac'] = 'H histidine'
cdn['caa'] = cdn['cag'] = 'Q glutamine'
cdn['cgt'] = cdn['cgc'] = cdn['cga'] = cdn['cgg'] = 'R arginine'
cdn['att'] = cdn['atc'] = cdn['ata'] = 'I isoleucine'
cdn['atg'] = 'M methionine'
cdn['act'] = cdn['acc'] = cdn['aca'] = cdn['acg'] = 'T threonine'
cdn['aat'] = cdn['aac'] = 'N asparagine'
cdn['aaa'] = cdn['aag'] = 'K lysine'
cdn['agt'] = cdn['agc'] = 'S serine'
cdn['aga'] = cdn['agg'] = 'R arginine'
cdn['gtt'] = cdn['gtc'] = cdn['gta'] = cdn['gtg'] = 'V valine'
cdn['gct'] = cdn['gcc'] = cdn['gca'] = cdn['gcg'] = 'A alanine'
cdn['gat'] = cdn['gac'] = 'D aspartic acid'
cdn['gaa'] = cdn['gag'] = 'E glutamic acid'
cdn['ggt'] = cdn['ggc'] = cdn['gga'] = cdn['ggg'] = 'G glycine'
For this question there is a warmup
problem.
Write a program that, starting from
the cdn dictionary above, generates a sort of inverse
dictionary. That is, it computes a new dictionary aacid,
such that aacid['glycine'] gives the
list ['ggt','ggc','gga','ggg'] and so on.
Submit the program
Variable names, values and id’s
There are some subtleties in python concerning variable names and their values, that may lead to nasty bugs if you are not aware of them.
As we saw in chapter 2 of ‘Hello World’, variables have names and values. The value is stored at a memory position, the identity of which can be accessed via:
>>>a=6
>>>id(a)
4298187248
Different names can refer to the same value at the same location:
>>>b=a
>>>print b
6
>>>id(b)
4298187248
In case immutable types are being changed, a new value is stored at a new position. If the old value was also attached to another name, it will not change and the old name will still be attached to it:
>>>a=7
>>>id(a)
4298187224
>>>print b
6
>>>id(b)
4298187248
Immutable types include int, float, and str. Immutable collections include tuples.
In contrast to immutable types, it is possible to change mutable types, while keeping the same identity. Thus, if the same value is attached to different names, the other names will also refer to the changed value:
>>>a=[6]
>>>b=a
>>>a[0]=7
>>>id(a)
4410098320
>>>id(b)
4410098320
>>>print b
[7]
If a variable is defined anew instead of being mutated, it will obtain another identity:
>>>a=[6]
>>>id(a)
4407452520
>>>b=a
>>>a=[7]
>>>id(a)
4410098320
>>>print b
[6]
>>>id(b)
4407452520
Mutable collections include lists, dictionaries and arrays.
Predict which values the parameters a and b will get after executing the following code:
a = [3,6,2]
b = a
b[2]=8
b=[3]
Predict which values the parameters a, b and c will refer to in the following case:
a = linspace(-1,1,5)
b = a
c = 1*a
b[0] = 26
This is a more complicated example. Predict which values a, b, c and d will refer to.
a=[3,2,4]
b=[7,a]
c=a[:]
d=b[:]
b[1][0]=10
b[0]=6
a[2]=50
In case you want to set one variable equal to another one, but you do not want them
to share any ids at any level anymore, the function copy.deepcopy() can be used:
import copy
a=[3,2,4]
b=[7,a]
d=copy.deepcopy(b)
b[1][0]=10
b[0]=6
a[2]=50
print 'a:',a
print 'b:',b
print 'd:',d
gives as output:
a: [10, 2, 50]
b: [6, [10, 2, 50]]
d: [7, [3, 2, 4]]
import copy
a=[3,2,4]
b=[7,copy.deepcopy(a)]
b[1][0]=10
b[0]=6
a[2]=50
print 'a:',a
print 'b:',b
gives as output:
a: [3, 2, 50]
b: [6, [10, 2, 4]]
List Comprehension
This section is optional.
List comprehension is a way of making a list using notation from
set theory. Here is an example.
N = 28
[n for n in range(1,N) if N % n == 0]
If the code looks mysterious at first, try looking at the output
and you will see what is going on.
Find the common biosequence names in brain and plasma
(see above) using list comprehension.
Gamow's Diamonds
This section is optional.
In the years between the structure of DNA being discovered and the
genetic code being correctly unravelled, there were several proposals
for what the genetic code could be. They are mostly forgotten now,
but one is still remembered, because though it was wrong, it was very
ingenious.
George Gamow guessed (correctly!) that codons of three bases would
map to 20 amino acids. Then he suggested that a sequence of bases
would form a sort of cage for an amino acid. Different codons would
give differently shaped cages, and each shape of cage would be just
right for amino acid.
Consider a codon, say ACG. On the other strand there would be
TGC.
Now consider the inclined `diamond' ACCG made from both strands.
This is what Gamow suggested might be a cage for an amino acid. Now,
the 43=64 possible codons correspond to 64 possible
diamonds. But some these are equivalent by symmetry. Tracing the
ACCG diamond backwards, we have AGCC, and starting from the other side
we have CCAG and CGAC:
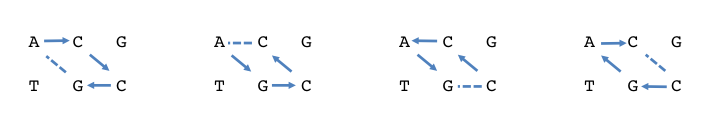 Show that allowing for the symmetries leaves exactly 20 different
Gamow diamonds.
Show that allowing for the symmetries leaves exactly 20 different
Gamow diamonds.
Submit the program